




Hirayama disease (HD) is a lower motor neurologic disorder that manifests in young males in their early 20s, manifesting with gradually progressive weakness and wasting of C7-T1 innervated muscles. Dynamic magnetic resonance imaging (MRI) clinches the diagnosis, and treatment is mainly supportive using a cervical brace, and in refractory cases, surgical. This review aims to describe HD pathogenesis, clinical features and management, and also throws light on the mimickers of HD.
Cervical flexion-induced myelopathy (CFIM), popularly called Hirayama disease, is a relatively uncommon neurologic disorder of the lower motor neurones initially described in Japan by Keizo Hirayama in 1959.1 Though originally called ‘juvenile muscular atrophy’, many other terms have been used to describe HD, such as benign juvenile brachial spinal muscular atrophy, monomelic amyotrophy, juvenile asymmetric segmental muscular atrophy, juvenile muscular atrophy of distal upper extremity and CFIM.2,3 Hirayama disease, coined in 1991, is the most commonly employed name.
As the names suggest, HD predominantly affects young males in their teens and early 20s. It manifests with gradually progressive weakness, atrophy and fasciculations of the hand and medial forearm muscles innervated by the C7 to T1 myotomes with brachioradialis sparing.1,2
The disease results from the forward displacement of the posterior dura on flexing the neck, leading to cord compression. Autopsy findings show that the main pathology is in the cervical anterior horn and anterior roots asymmetrically.4 Hence, treatment is aimed at avoiding neck flexion, which is achieved by a hard cervical collar, and if intractable, with surgery.5,6 There is a lack of knowledge of HD among clinicians due to its relative rarity, and this review aims to improve awareness to enable early diagnosis and treatment, thereby preventing disability progression.
History and epidemiology
Japanese neurologist Keizo Hirayama reported the first patients with HD in 1959. He reported 12 patients and named their condition ‘juvenile muscular atrophy of the unilateral upper extremity’.1 In 1982, it was hypothesized that HD results from lower cervical cord compression due to disproportionate growth of the spinal cord and vertebral canal.7 The first autopsy and pathologic study was performed in 1987, which found extensive anterior horn cell shrinkage in the C7 and C8 regions.4
A Japanese epidemiologic survey in 2006 contained details on epidemiology, illness progression, imaging, electromyographic findings and treatment of HD, and is the most extensive survey of patients with HD to date.8 The survey identified 333 cases of HD. The peak age of onset was 15–17 years, with a striking male predominance, a slow onset and progression, and stabilization in ≤6 years. The survey observed a predominantly unilateral upper limb involvement, worsened by exposure to cold. Two familial cases were also described.
HD is primarily seen in the Asian population, mainly from Japan, China, India, Sri Lanka and Taiwan.8–12 However, there are reports from Australia, Europe and North America.13–15 The reason for the high incidence in Asian countries is still unexplained; ethnic and geographic factors might be responsible and need further evaluation. The male-to-female ratio varies considerably between countries, with a strong male predominance (2.8:1 in India).16
The disease affects patients aged 15–25 years.8 Hirayama noted that the peak age of disease onset was 2 years after the longitudinal growth curve peak of adolescents.1 He thus proposed that an imbalance in growth between the spinal column and the dural sac during the pubertal growth spurt was responsible for the disease. The male preponderance was explained by the more rapid height increase in males compared with females during puberty.1 HD is not a hereditary condition; however, a few familial cases of HD have been reported.17
The proposed pathophysiologic mechanisms in Hirayama disease
Initial studies described HD as a variant of spinal muscular atrophy (SMA), since the clinical features were similar between the conditions.18 However, unlike those with SMA, genetic analysis of patients with HD did not reveal survival of motor neuron (SMN) gene deletion, and they had a comparatively good prognosis.19
The possible pathophysiologic mechanisms underlying HD include:
- Dysplasia of the dural sac. Hirayama et al. proposed that during the pubertal growth spurt, there is an imbalance between the growth of the spinal cord and spinal canal, contributing to the tightness of the dura.1 On cervical flexion, anterior dural displacement occurs leading to impingement of the spinal cord against the bony vertebrae.20 Compression of the anterior spinal artery territory causes ischaemia, leading to anterior horn cell infarction of in the C7–T1 areas.21–24
- Nerve root dysplasia. Because of nerve root dysplasia, on neck extension, the posterior roots of the involved side become less slack. However, on neck flexion, the posterior roots are unable to extend, which displaces the lower cervical cord anteriorly and laterally on the involved side.25
- Spinal ligament abnormalities. Loss of the elastic fibres of the spinal ligament, loss of the epidural ligament and the normal wavy structure of the dura lead to tightness of the posterior dura.26,27
- Engorged posterior epidural venous plexus. The forward dural shift creates a negative pressure in the posterior epidural space, impairing venous return during flexion of neck. This leads to blood being diverted into the posterior epidural plexus, which causes ischaemia in the anterior horn cells and ventral nerve roots in the cervical cord.28 This is the latest postulated pathophysiologic mechanism in HD, leading to focal ischaemic poliomyelopathy.4
- Immunologic mechanisms. Kira and Ochi noted a high frequency of atopy and elevated immunoglobulin (Ig)E levels in patients with HD.29 As well, Ito et al. found that severe clinical manifestations were seen in patients with elevated serum IgE levels.30 Others proposed that IgA deficiency might predispose patients to developing HD.31 Anti-GQ1b antibody was also proposed as a prognostic factor; however, all such studies were retrospective and had a small sample size.32 Therefore, these immunologic factors cannot be considered causes of HD until confirmed by large-scale prospective studies.
In addition to the above factors, Khadilkar et al. demonstrated that individuals with HD had longer necks compared with controls.33 Also, an imbalance between the strength of neck flexors and neck extensors causes instability and predisposes patients to spinal cord atrophy.33,34 Dynamic instability was explained by Wang et al. by demonstrating the variations in the neutral and flexion Cobb angles on cervical X-rays in patients with HD.35 Lastly, Biondi et al. proposed that repeated subclinical neck trauma contributes to microcirculatory disturbances during physical exertion that predisposes patients to cord damage.36
Familial cases of Hirayama disease
Though HD is a sporadic condition, a few familial cases have been reported.37 Misra et al. described 15 males with HD from 14 Indian families; in this cohort, two patients were brothers.19
Atchayaram et al. reported a mother and son with HD.17 The disease in the son manifested at age 21 years, with gradually progressive amyotrophy of the left forearm and intrinsic hand muscles lasting 3 years. He also had fasciculations, tremors and cold-induced paresis. The patient’s mother had also developed cold-induced paresis in the right hand at 20 years of age, followed by hand and forearm weakness 10 years later. Brachioradialis was spared in both patients. Electrophysiology in both patients revealed chronic denervation with reinnervation. MRI of the mother’s cervical spine showed signal changes at C4–C5, but was normal in the son.17
In other cases, Pandey and Jain reported a father and daughter with bilateral HD,38 and Sobue et al. reported an affected father and a son.39 Schlegel et al. described a Caucasian father and son with HD-like presentation characterized as focal amyotrophy of the upper limb, with onset at the age of 16 in both, suggesting autosomal dominant inheritance.40 Hirayama also reported familial cases of HD.41
Another case series reported a Greek family with four members over three generations who presented with distal limb amyotrophy consistent with HD, suggesting an autosomal dominant mode of inheritance.42 This is the first case series describing three generations affected by HD.
Genes and Hirayama disease pending confirmation
Two variant genes are associated with HD and have been identified by whole-exome sequencing: KIAA1377 (also known as Centrosomal Protein 126 [CEP 126]) and chromosome 5 open reading frame 42 (C5orf42).43 These two genes were identified in four Korean male patients with monomelic amyotrophy, and then by selective genotyping of candidate variants in 24 patients. A G668S (glycine to serine) variant in the CEP 126 gene on chromosome 11q22, and a P1794L (proline to leucine) variant in the C5orf42 gene on chromosome 5p13 were identified. Four patients with the G668S variant carried it on the same haplotype, suggesting a common origin. Having only a single copy of the causal variant in either gene did not seem to promote risk for developing monomelic amyotrophy. A combination of heterozygosity for both variants or with homozygosity for the CEP 126 variant was significantly associated with disease development. This suggested that the CEP 126 and C5orf42 genes may synergistically act as susceptibility genes for developing monomelic amyotrophy.43
Pathology in Hirayama disease
The first autopsy was performed by Hirayama in a 38-year-old male (after he died of lung cancer) who had left upper limb distal weakness, tremulousness, atrophy and cold-induced worsening, beginning at the age of 15 years and progressing for 1 year.4 Examination revealed anteroposterior flattening of the spinal cord, and histopathologic findings included significantly smaller bilateral anterior horns at C7 and C8, which were more towards the left. The lesions showed central necrosis without cavitation, degenerative changes and astrocytic gliosis. There was no evidence of infiltration by mononuclear phagocytes. The dorsal horns and the posterior nerve roots were spared. These changes were postulated to be related to circulatory insufficiency and ischaemia, which is how HD came to be known as focal ischaemic cervical poliomyelopathy.44
Disease manifestations
Salient features
HD has five salient features, which have been described by Tashiro and others:8
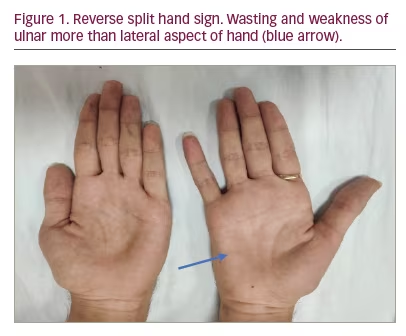
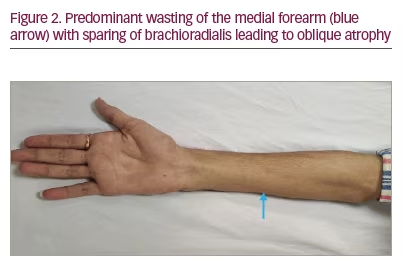
- Distal weakness with wasting, predominantly to the medial aspect of one upper limb or both upper limbs asymmetrically. Predominant wasting of the ulnar part of hand leads to the ‘reverse split hand sign’ (Figure 1) that differentiates HD from amyotrophic lateral sclerosis (ALS); this occurs because the anterior horn cells that innervate the ulnar aspect of the upper limb are preferentially affected. Also, preferential medial forearm involvement with the sparing of brachioradialis leads to ‘oblique amyotrophy’ (Figure 2).
- Insidious onset between age 10 years to early 20s with gradual progression over 3–5 years and followed by quiescence.
- Tremors, which are irregular and coarse in the affected fingers, with polyminimyoclonus.
- Transient symptomatic worsening in cold climate. This is attributed to on-going denervation blocking conduction of the muscle fibre membrane in reinnervating muscles.45
- No objective loss of sensation and electromyographic changes (EMG) evidence of chronic denervation in affected or subclinically involved muscles.
Atypical features
HD also may manifest with some atypical features as given below:
- Pyramidal signs. Hyper-reflexia and inverted reflexes can be a feature in some patients due to severe cord involvement.46 Pyramidal signs were reported in 2.4% and 10.6% of patients with HD in Japan and China, respectively.8,47 Upper motor neurone involvement has also been noted in 18.0% and 12.0% of the patients reported by Hassan et al. and Sonwalkar et al., respectively.48,49 Upper motor neurone signs imply severe cord involvement and indicate a worse outcome.50
- Proximal upper limb weakness and atrophy. The lower cervical cord C7–T1 is mainly affected in HD, hence the atrophy usually involves the distal upper limbs. However, a retrospective cohort study reported pure proximal involvement of the upper limb in 3.6% of patients and symptomatic/asymptomatic proximal wasting in approximately 40.0% of patients.10 Hirayama et al. also reported two cases (out of 20) with proximal involvement: one each with pectoral and triceps weakness.5
- Sensory involvement. Sensory tract involvement due to compressive myelopathy can cause sensory deficits in some patients with HD. Tashiro et al. reported that 19.2% of patients had sensory impairment.8
- Long disease progression. Most patients progress over 3–5 years, and the disease plateaus after that. However, there have been reports of disease progression beyond 10 years.28
In the absence of definitive guidelines, a group has put forward clinician-led guidelines on diagnosing and treating HD. They include most of the above symptoms that should be considered in the diagnosis of HD.51
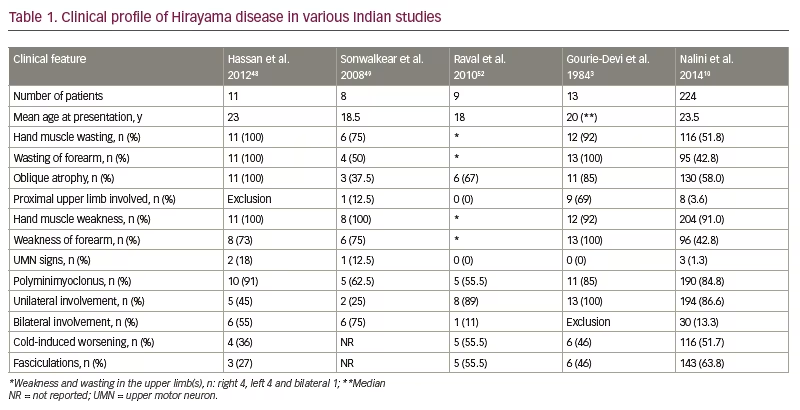
The clinical profile of HD in various studies conducted in India over the last 20 years is given in Table 1.3,10,48,49,52
Neuroimaging findings
The most important diagnostic tool in evaluating a patient with suspected HD is MRI of the cervical spine in two positions: neutral and flexion.49,52 This type of imaging is known as a dynamic MRI. The image acquisition is performed on a 1.5 T or 3.0 T scanner in supine position. The scanning involves the following sequences: sagittal and axial T1-weighted sections, sagittal and axial T2-weighted sections, and sagittal and axial T1-weighted images with fat saturation with contrast.
Neutral magnetic resonance imaging of the cervical spine
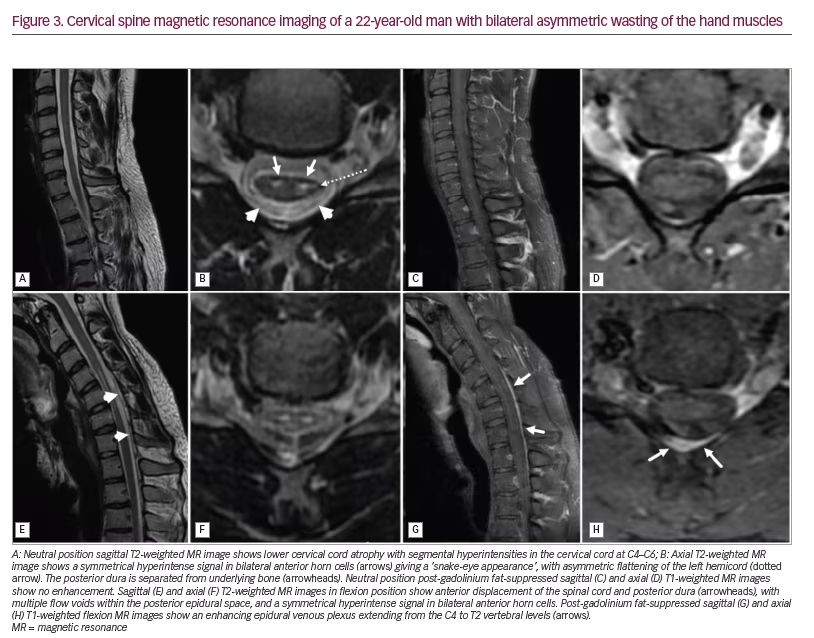
Many salient features of a neutral scan enable the radiologist to pick up HD. The characteristic findings on neutral scans include loss of normal cervical lordosis or abnormal curvature of the spine, loss of posterior attachment of dura to the subjacent lamina, localized lower cervical cord atrophy, asymmetric flattening of cord anteroposteriorly, and intramedullary T2 cord hyperintensity (Figure 3A–D). Chen and colleagues have demonstrated the sensitivity and specificity of findings on neutral cervical spine images.53 Among the MRI features, cord thinning, cord flattening and loss of dural attachment (LOA) to the lamina were found to have more than 80% accuracy in identifying disease. The LOA sign was the most valuable finding on neutral MRI, with a sensitivity of 94.0% and specificity of 98.0%.53
In some patients, on axial sections, an asymmetrical bilateral T2 hyperintense signal may be appreciated; this has been described as the ‘snake-eye appearance’, as it resembles a snake’s face (Figure 3B).54 Also, this sign suggests a severe lesion and the need for timely surgery.55
Flexion magnetic resonance imaging of the cervical spine
Cervical flexion MRI is the most important investigation, as it confirms the diagnosis in all suspected HD cases. It reveals HD pathogenesis and corresponds to the clinical manifestations. An angle of 35° flexion of the neck is required for an adequate scan.20 The salient radiologic signs include forward displacement of the posterior dura, asymmetric lower cervical cord flattening, cord atrophy, epidural flow voids and a high-intensity crescent-shaped mass (Figure 3E–H). Another vital sign that can be appreciated is the anterior cord displacement. The vertebral canal–dural sac growth imbalance, and the dura attached only at C2–C3 and coccyx, leads to dural sac tightness on cervical flexion. This displaces the dura and the spinal cord forwards during neck flexion. Repeated neck flexion also causes chronic microcirculatory changes, which can affect the anterior horn cells and lead to cord thinning. The crescent-shaped mass on neck flexion occurs because of the congestion of the posterior epidural venous plexus, and the mass disappears on neutral imaging. A contrast study reveals enhancement of the epidural venous plexus (Figure 3G and 3H).52,53,56 In a cohort study, dynamic MRI was noted to have a sensitivity of 71% and specificity of 100% in diagnosing HD.56
Role of functional imaging
Diffusion tensor imaging (DTI), which is employed in HD diagnosis, uses the principle of Brownian motion of water particles.57 An increased apparent diffusion coefficient and lower fractional anisotropy are potential clues.58 DTI can not only evaluate the cord injury but also guide the level at which surgery must be performed. A study of 10 patients with HD reported the corticospinal tract DTI changes in the brain, and tried to correlate them with clinical findings.59
Functional imaging is an important consideration for future research. Functional MRI employing the blood oxygen level-dependent technique was studied in 17 patients with HD who underwent cervical decompression with fusion surgery.60 Patients were imaged before surgery and then at different intervals post-operatively. The study revealed ipsilateral motor cortex activation when the affected hand was used compared with the unaffected hand. The activation was reduced following surgical fixation.60
Electrophysiology of Hirayama disease
Electrophysiologic studies are critical in supporting the diagnosis and differentiating various HD mimics. The EMG changes are confined to the C7–T1 myotomes.61 The characteristic EMG findings include fibrillations and positive sharp waves in those with on-going denervation, and large amplitude, long duration, polyphasic, motor unit action potentials with moderate to discrete recruitment in patients with long-standing disease.62
Motor nerve conduction studies may be normal or show reduced compound muscle action potential (CMAP) amplitudes in the affected upper extremities, with normal conduction velocities.48 The ulnar/median (U/M) CMAP ratio can be employed in differentiating HD from ALS and cervical spondylotic myelopathy. The U/M CMAP ratio is decreased in patients with HD, higher in patients with ALS, and normal in patients with spondylosis.63 Sensory conduction is normal in patients with HD.61,62
The F waves show reduced frequency and conduction velocity with normal latency in patients with HD.61 One study reported normal F-wave velocities, while the other reported prolonged F-wave latency in HD.48,64 Repeater F waves were also commonly noted in the median and ulnar nerve motor studies.58
Somatosensory evoked potential (SEP) and motor evoked potential (MEP) changes have also been documented in HD. Abraham et al. demonstrated normal SEPs in patients with HD.65 Park et al. studied SEP parameters during neutral and cervical flexion positions in patients with HD and controls, and demonstrated changes in the N9-N13 and N13-N20 interpeak latencies.66 In the HD cohort, they found a significant difference in the mean N13-N20 interpeak latency during cervical flexion, and it correlated strongly with the presence of HD.66 Changes in the MEP have also been shown in neck flexion, such as reduced amplitude, longer central motor conduction time and prolonged latency.65,67 Another study found that the intraoperative MEP amplitude increased during surgery and persisted until surgery completion.68 Thus, the MEP amplitude may help define an adequate position for cervical fixation.68
Motor unit number estimation (MUNE) estimates the number of motor units supplying a muscle, and can be used to measure the severity and progress of HD, along with treatment effect.69 A reduced MUNE value in a subclinically affected muscle suggests that the disease has started on the asymptomatic side, and therefore, measures to halt its progression need to be used. The motor unit number index can also show subclinical changes before muscle power is affected.69
Updated diagnostic criteria of Hirayama disease
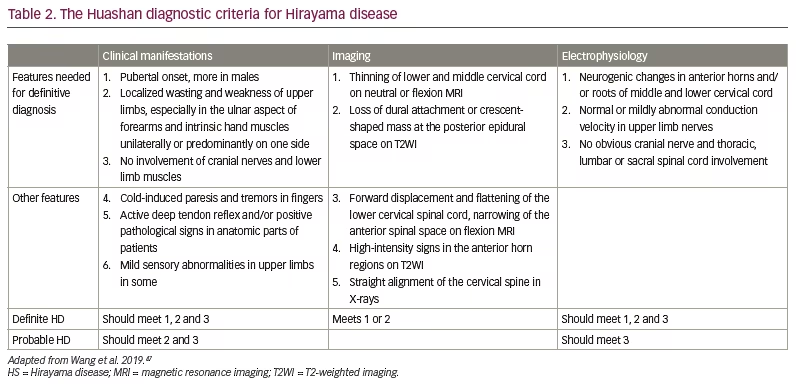
The definitive diagnosis of HD requires EMG and neck flexion MRI. The revised criteria consider the clinical manifestations, MRI characteristics and electrophysiology findings.47 These criteria do not mention ‘progression of the disease over 3–5 years’, which was put forward initially by Hirayama.44 They also do not emphasize the absence of sensory deficits and or pyramidal features. The updated diagnostic criteria (Huashan criteria) are given in Table 2.47
Hirayama disease mimics
The following conditions can present with weakness and atrophy of upper limb muscles similar to HD, and need to be considered in the differential diagnoses:
- Early-onset ALS
- Flail arm variant of motor neurone disease
- Distal SMA
- Cervical spondylotic myelopathy
- Syringomyelia
- Ossification of posterior longitudinal ligament of the cervical spine
- Intramedullary tumours
- Peripheral nerve compression syndromes (e.g. carpal tunnel syndrome, thoracic outlet syndrome)
- Hereditary motor neuropathies with upper limb onset.
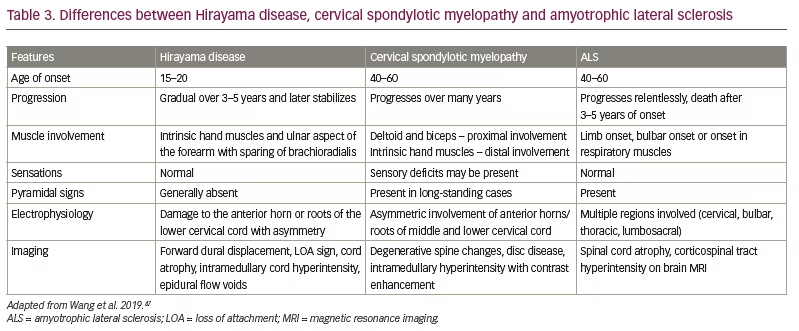
Structural lesions can be picked up on cervical spine MRI or computed tomography; nerve conduction studies will help diagnose peripheral nerve compression disorders. However, ALS and cervical spondylotic myelopathy can mimic HD in the early stages. Table 3 shows the major differences between ALS, HD and cervical spondylotic myelopathy.47
Current treatment aspects
Non-surgical management
There is no consensus regarding the management of HD, and no drugs are effective in treating HD. Although the disease is self-limiting in most patients, the disability produced is usually significant. Therefore, measures to halt disease progression must be employed before significant disability is established. Applying a hard cervical collar is the first line and most routinely used treatment.45 It prevents disease progression by restricting neck movements, which avoids cervical cord compression.45
Two studies reported improved strength of affected muscles after applying a cervical collar.70,71 Tokumaru et al. evaluated the therapeutic benefits of collars in 38 patients with HD and compared them with 45 untreated patients.5 The disease duration decreased after applying the cervical collar; 15 patients also experienced an improvement in weakness and cold paresis, suggesting its efficacy. Narayana et al. also demonstrated the utility of the collar in HD.72 However, using cervical collars for 24 hours a day for 3–4 years is usually cumbersome and unbearable for many patients.73
The reason for the improved strength of muscles after a conservative approach is possibly reinnervation.74 Exercise therapy to improve neck muscle strength has been widely employed and is reported to be beneficial.75
Neurosurgical management
The various interventions are based on pathogenesis theories. The indications for surgical intervention include a rapid progression of severe disease, positive pyramidal signs, failure of conservative treatment and poor compliance to the neck collar.6
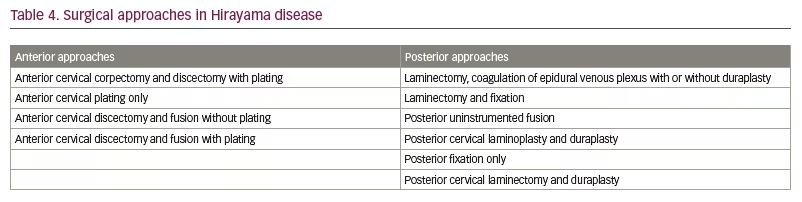
There are many anterior and posterior neck approaches described in various studies. The surgical approaches described in the literature include laminectomy, duraplasty, corpectomy, discectomy, decompression and spinal fusion (Table 4).6 The superiority of one technique over the other has not been convincingly proved. Surgery aims to avoid further cord compression by preventing neck flexion. Possible benefits of surgery include shortening the natural history of the disease, preventing recurrence of venous congestion and improving the hand grip strength.13,76
Early surgery may preserve functionally inhibited motor units and may avoid progressive deterioration of existing motor units. Surgery also reverses the abnormal F-wave changes and MEP, along with improving imaging.68,76,77
A meta-analysis on the impact of various surgical procedures in patients with HD described 10 neurosurgical procedures; the most commonly performed was anterior cervical discectomy and fusion (ACDF) followed by plating.6 The pooled proportion of patients who improved following all approaches was 80.0% (95% confidence interval [CI] 76–84%). It was highest for anterior cervical plating (96% [95% CI 62–100%]) and lowest for ACDF without plating (57.0% [95% CI 20–88%]).6 A definitive answer on the efficacy of surgical procedures requires a high-quality randomized controlled trial.
The Huashan classification system guides us in the management of HD:47
Type 1: Atrophy of hand and forearm muscles or asymmetric bilateral atrophy in upper limbs with Type 1a (stable) and Type 1b (progressive).
Type 2: Atrophy with pyramidal involvement.
Type 3: Atypical with atrophy of proximal upper limb muscle or symmetric upper limbs or presence of sensory dysfunction.
Type 1 and Type 3 HD are managed conservatively, while Type 2 disease requires surgical intervention.
Prognosis
HD is a predominantly self-limiting disease. It progresses for 3–5 years and later stops progressing. Cervical collar treatment improved 57.2% of patients.8 Moreover, 46.3–87.6% of patients who underwent surgery had good outcomes at follow-up.6 However, the patients who develop spasticity or other pyramidal signs, and those with a snake-eye appearance on MRI, tend to have poor outcomes.55
Conclusion
HD is a rare disease that leads to weakness and atrophy of unilateral or bilateral hand muscles, commonly encountered in young Asian males. Dynamic MRI confirms the diagnosis in a clinically suspected patient. Treatment mainly aims to reduce neck flexion; therefore, long-term cervical braces are the mainstay. Surgical treatment is employed in patients who progress with conservative treatment, but the best approach is still debated. Further studies are needed, including genetic studies to unravel the exact HD pathogenesis in order to develop more effective treatments.


