




Myasthenia gravis (MG) represents the major and most frequent primary disorder of the neuromuscular junction. Clinical hallmarks are variable and include exercise-induced weakness involving extraocular, bulbar, limb and/or axial muscles.1 Respiratory involvement, characterized by orthopnoea or dyspnoea with exertion, occurs in about 40% of patients with MG and is often associated with bulbar dysfunction and neck weakness. Most frequently, extraocular muscle involvement, with typical asymmetrical ptosis and/or diplopia, is the first symptom to appear. Within 3 years, only 20% of patients have disease that remains confined to extraocular muscles; most patients progress to generalized MG (gMG).1
About 85–90% of patients with gMG display pathogenic immunoglobulin (Ig)G antibodies, which are most often directed against the skeletal muscle nicotinic acetylcholine receptor (AChR) and less frequently against the muscle-specific tyrosine kinase (MUSK) and the lipoprotein receptor-related protein (LRP4). A small percentage of patients, however, are defined as seronegative, as they do not have antibodies identifiable with current techniques. The general pathogenic effect of these autoantibodies is disrupted neuromuscular transmission, which translates clinically into the typical fatigability and weakness of MG.1 Anti-AChR antibodies are mainly of the IgG1 and IgG3 subclass, and can therefore bind and activate the complement cascade, which leads to the destruction of the post-synaptic folds.2 Other important pathogenic mechanisms of anti-AChR antibodies are direct blocking of the acetylcholine binding site on the AChR3 and crosslinking of AChR with its subsequent internalization and degradation.4
About 40% of anti-AChR seronegative patients display MUSK antibodies. These antibodies are of the IgG4 subclass and have limited ability to activate the complement system. Instead, they block AChR clustering, which impairs neuromuscular transmission.5 Similarly, anti-LRP4 antibodies, which are present in about 19% of double-seronegative patients, block AChR clustering by inhibiting the agrin–LRP4 interaction needed to activate MUSK.6–8 Furthermore, anti-LRP4 antibodies can also activate the complement cascade, as they belong mainly to the IgG1 subclass.9
Taking into account epidemiology, autoantibodies, clinical presentation and comorbidities, it is possible to stratify MG into subgroups with different therapeutic and prognostic implications. AChR-seropositive (AChR+) MG can be divided into early-onset MG (EOMG), when onset of symptoms is before age 50 years, and late-onset MG (LOMG), when onset of symptoms is after age 50 years.10 EOMG is three times more frequent in females, and is associated with the human leukocyte antigen (HLA) genes HLA-DR3 and HLA-B8.11,12 EOMG often presents with thymic follicular hyperplasia, although it is not a prerequisite, and responds well to thymectomy.10 On the other hand, LOMG is slightly more frequent in males and is weakly associated with the HLA-DR2, HLA-B7 and HLA-DRB1*15:01 genes.13 These patients only rarely present with thymic hyperplasia and generally do not respond to thymectomy.14 The international consensus on managing MG recommends early thymectomy in non-thymomatous, AChR+ gMG only in patients aged 18–50 years.15 Patients with thymoma, regardless of their age, are considered as a separate subgroup. Thymoma-associated MG is a paraneoplastic disorder in which nearly all patients display anti-AChR antibodies and generalized disease. Generally, 40% of patients with thymoma develop MG and even more can present with anti-AChR antibodies without MG.16 MUSK-associated MG generally affects adults, sparing the elderly and children,17 and is not associated with thymic alterations (though sporadic cases have been diagnosed with thymoma).18 Thymectomy is not recommended in MUSK-seropositive (MUSK+) MG.15 MUSK-associated MG is associated with the HLA-DQ5 gene and predominantly involves the cranial and bulbar muscles.19–21 LRP4-associated MG generally presents with mild gGM or ocular MG. The thymus is atrophic in two-thirds of patients, but thymic hyperplasia has also been described.6
Besides symptomatic treatment with anti-cholinesterases, which induce mild to moderate side effects and do not generally lead to pharmacological remission, current MG therapy is based either on immunomodulation or non-specific immunosuppression. Immunomodulation with intravenous immune globulin (IVIg) and plasmapheresis is mainly recommended as first-line treatment for MG exacerbation or crisis, yet many patients who do not respond to conventional immunosuppression benefit from chronic treatment with either IVIg or plasmapheresis.15,22 Immunosuppression includes corticosteroids and non-steroidal immunosuppressive therapies (NSISTs), such as calcineurin inhibitors (e.g. cyclosporine and tacrolimus) and anti-metabolites (e.g. azathioprine, mycophenolate mofetil, methotrexate or cyclophosphamide). NSISTs can be used either in combination with oral steroids or as monotherapy. However, due to their delayed response, bridging with prednisone is generally recommended until peak efficacy is obtained.10
Both steroids and NSISTs suppress the immune system in a non-selective manner, and their use is often limited by their wide range of side effects. In particular, prolonged use of prednisone can induce many debilitating side effects, such as osteoporosis, high arterial blood pressure, glaucoma, diabetes, weight gain, irritability and skin atrophy, which can greatly impact quality of life.23 Furthermore, 10–20% of patients with MG are refractory to conventional immunosuppression.24 Poor tolerance and the relatively high incidence of refractoriness highlight the need for improved MG therapies, and more research is needed to develop a more targeted and well-tolerated therapy, as is the case for many autoimmune disorders.
Recently, research has flourished in new therapeutic molecules with a more specific effect.25 Efgartigimod (formerly ARGX-113), a humanized IgG1-derived fragment crystallizable (Fc) region that competitively blocks the neonatal Fc receptor (FcRn), is a new drug that has demonstrated good safety and clinical efficacy in a phase III trial.26 Following such encouraging results, in December 2021, the US Food and Drug Administration (FDA) approved efgartigimod for the treatment of patients with AChR+ gMG, making it the first FDA-approved FcRn antagonist, leading the way to a new and innovative treatment option for MG. This review describes the clinical development and mode of action of efgartigimod, which represents a encouraging new therapy for gMG.27
Efgartigimod
Physiological function of the neonatal fragment crystallizable receptor
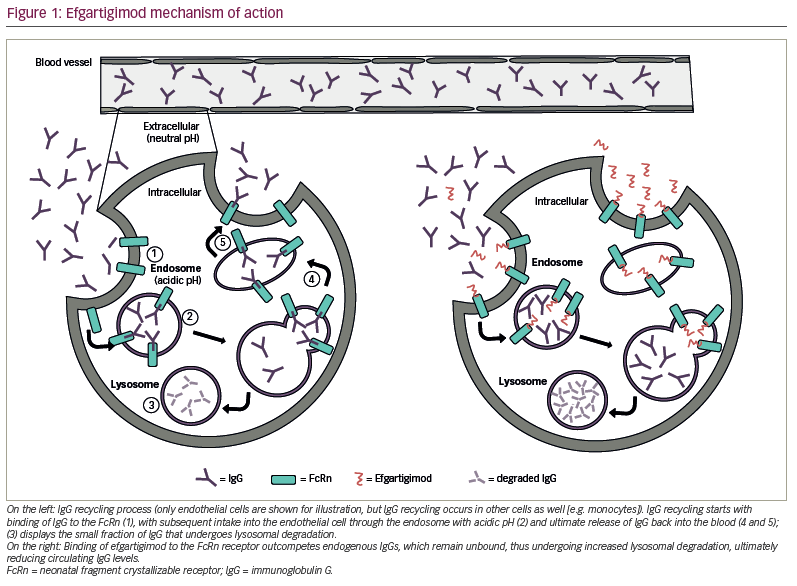
The FcRn is a major histocompatibility complex I class-like molecule that is almost ubiquitously expressed and functions as a recycling and transcytosis receptor for IgG and albumin, which are the most abundant serum proteins. IgG is salvaged by FcRn from intracellular lysosomal degradation via an acidic pH-dependent cellular recycling process, since FcRn is able to bind IgG only at mildly acidic pH (5.0–6.5).28 IgG and albumin are internalized through pinocytosis in an acidic endosomal compartment, where they are retained by FcRn binding, thus escaping lysosomal degradation. The endosome then reaches the plasma membrane where its contents are exocytosed at physiological pH. This cellular transport mechanism is at the base of the recycling and increased half-life of albumin and IgG, which, compared with other immunoglobulins such as IgA or IgM, has a fourfold longer half-life.28,29
FcRn function varies according to its tissue expression. For example, FcRn expression on the placenta allows transfer of IgG from the mother to the foetus, ensuring the immune protection of the newborn.30 In the intestine, there is a bidirectional transcytosis, with delivery of IgG into the lumen but also transcytosis of IgG or IgG immune complexes back into the lamina propria, with the scope of delivering luminal antigens in the form of IgG immune complexes to mucosal dendritic cells, which then manage immune responses.31 In the kidneys, FcRn is expressed on podocytes, clearing IgG by transferring it from the filtration membrane to the urinary space. This prevents IgG and IgG immune complexes accumulating, which could lead to organ damage.32 Moreover, FcRn expressed on the proximal tubule epithelial cells is involved in albumin reabsorption.33 In the central nervous system (CNS), FcRn is expressed on the choroid plexus epithelium and microvascular endothelium, where it removes IgG by reverse transcytosis from the CNS back into the circulation, a process that may limit CNS inflammation in pathological situations (e.g. bacteraemia).34
Pharmacodynamics
While the importance of the FcRn–IgG interaction is undisputed in normal physiology, in autoimmune disorders, characterized by pathogenic self-reactive IgG antibodies, the FcRn-mediated recycling process perpetuates the harmful effects of such pathogenic IgG. Therefore, decreasing the circulating levels of self-reactive IgG could be beneficial.
Efgartigimod is a human IgG1-derived Fc fragment that binds to the human FcRn with nanomolar affinity. It has been modified exploiting the novel ABDEG™ technology, which dramatically increases its affinity for FcRn at both physiological and acidic pH, thus allowing it to outcompete endogenous IgG binding.35 The result is a constitutive blockage of FcRn-mediated recycling of IgG with subsequently increased IgG degradation (Figure 1).36 Intravenous (IV) administration of efgartigimod has been reported to reduce IgG levels in cynomolgus monkeys and in a mouse model of MG.36,37 Moreover, in a mouse model of MUSK+ MG, efgartigimod-treated mice had improved muscle weakness and fatigability compared with control littermates.37 In a study of 62 healthy volunteers, multiple doses of efgartigimod decreased IgG serum levels by approximately 75% on average and, in some cases, up to 85%.36 Serum levels of other immunoglobulins (e.g. IgA or IgM) or albumin were not reduced after efgartigimod administration, while total IgG levels returned to baseline approximately 8 weeks after the last administration.36 Similarly, in patients with gMG, both total IgG levels and AChR-antibody levels rapidly decreased after IV administration of efgartigimod.26 Total IgG serum levels have also been reported to be reduced in other autoimmune disorders, including immune thrombocytopenia and pemphigus vulgaris and foliaceous.38,39 In an open-label phase I study with healthy volunteers, initial IV administration of efgartigimod followed by weekly subcutaneous administration of 300 mg was able to keep total IgG serum levels reduced by approximately 50% at steady state.40
Pharmacokinetics
The recommended treatment cycle of efgartigimod is 10 mg/kg administered IV over 1 hour once a week for 4 weeks. For patients weighing ≥120 kg, the recommended dose is 1,200 mg per infusion.41 Subsequent treatment cycles can be given based on individual clinical response, at least 8 weeks after the previous cycle. Efgartigimod has linear pharmacokinetics, a volume of distribution of 15–20 L and a terminal half-life of 80–120 hours (3–5 days).41 A phase I open-label study of subcutaneous efgartigimod in healthy participants demonstrated a similar half-life to IV infusion.40
Efgartigimod is degraded by proteolytic enzymes into small peptides and amino acids. After a single IV injection of efgartigimod (10 mg/kg) in healthy participants, less than 0.1% of the dose was recovered in urine.41 No dedicated pharmacokinetic studies have been conducted in patients with hepatic impairment. In case of mild renal impairment, no dose adjustment is needed, while insufficient data are available to evaluate the impact of moderate (estimated glomerular filtration rate [eGFR] 30–59 mL/min/1.73 m²) and severe (eGFR <30 mL/min/1.73 m²) renal failure on efgartigimod pharmacokinetics.
Since efgartigimod is not metabolized by cytochrome P450, it does not interfere with drugs that are inducers, inhibitors or substrates of cytochrome P450.41 On the other hand, it is expected to reduce the systemic exposure, and therefore the effectiveness, of drugs that bind to the FcRn (e.g. immunoglobulin products, monoclonal antibodies or antibody derivatives containing the human Fc domain of the IgG subclass).41
Clinical trials in myasthenia gravis
A phase II, exploratory, randomized, double-blind, placebo-controlled study provided class I evidence that efgartigimod is safe and well tolerated in patients with gMG.42 Indeed, all patients in the efgartigimod arm displayed a rapid decrease in total IgG and AChR-antibody levels, without impacting other immunoglobulins or albumin levels. These reductions were accompanied by rapid, clinically meaningful and long-lasting disease improvement in 75% of cases.42
The recently completed ADAPT study (An efficacy and safety study of ARGX-113 in patients with myasthenia gravis who have generalized muscle weakness [ADAPT]; ClinicalTrials.gov identifier: NCT03669588) was a phase III, multicentre, randomized, double-blind, placebo-controlled trial evaluating the safety, efficacy and tolerability of efgartigimod in patients with gMG.26 The study included 167 patients aged at least 18 years with gMG, with or without AChR antibodies, with disease categorized between Myasthenia Gravis Foundation of America class II and class IVb and a Myasthenia Gravis Activities of Daily Living (MG-ADL) score of at least 5 (with >50% of the MG-ADL score due to non-ocular symptoms). Patients had to be on a stable dose of at least one MG treatment (e.g. anti-cholinesterases, oral steroids or NSISTs) and were randomized 1:1 to receive either IV efgartigimod 10 mg/kg or placebo, with weekly infusions over 4 weeks per cycle. Based on individual clinical evaluation, patients could receive subsequent cycles with an interval of at least 8 weeks from the previous cycle. Patients who participated could enter the open-label extension (OLE) ADAPT+ trial (A safety and tolerability study of ARGX-113 in patients with myasthenia gravis who have generalized muscle weakness [ADAPT+]; ClinicalTrials.gov identifier: NCT03770403), which is currently on-going.43
The primary efficacy endpoint was the proportion of patients with AChR+ gMG who were MG-ADL responders in the first treatment cycle, where response was defined as at least a 2-point reduction in the MG-ADL score sustained for at least 4 consecutive weeks, with the first improvement occurring 1 week after the first cycle.26 Among the 167 patients enrolled, 129 (77%) were AChR+ and, of these, 65 were in the efgartigimod group and 64 in the placebo group. In the AChR+ gMG efgartigimod group, a significantly higher proportion of patients (68%) were MG-ADL responders, meeting the primary endpoint, compared with patients in the placebo group (19%; p<0.0001). Similarly, compared with nine patients in the placebo group, 41 AChR+ patients in the efgartigimod group were Quantitative Myasthenia Gravis (QMG) responders, which was defined as a reduction of at least 3 points in the QMG score for at least 4 consecutive weeks after the first treatment cycle. Furthermore, patients enrolled in the efgartigimod group experienced a greater improvement in the Myasthenia Gravis Composite score and the Myasthenia
Gravis-Specific Quality of Life 15-item scale (MG-QOL15r) questionnaire.26
After a second cycle, the number of MG-ADL responders further increased to 71% in the efgartigimod group, compared with only 26% in the placebo group. Clinical improvement corresponded to a reduction of total IgG and AChR-antibody serum levels. Comparable results were also observed in the AChR seronegative population, with a clinically meaningful reduction in all assessment scores and total IgG serum levels in the efgartigimod-treated group. Efgartigimod was well tolerated and had a good safety profile. The most common adverse events were headache, upper respiratory tract infections and urinary tract infections. The severity of infections was generally mild or moderate.26
Currently, an expanded-access programme for efgartigimod is available for both AChR+ and AChR- patients with gMG who cannot enter a clinical trial, before its regulatory.44 On 17 December 2021 the FDA approved efgartigimod for the treatment of AChR+ gMG, and it is now sold under the brand name Vyvgart® (Argenx, Boston, MA, USA). Efgartigimod has thus become the first and only FDA-approved FcRn inhibitor.27 On 20 January 2022, efgartigimod was also approved in Japan for the treatment of patients with gMG, regardless of their antibody status.45
A phase IIIb, randomized, open-label study assessing the efficacy, safety and tolerability of a continuous IV efgartigimod regimen compared with a cyclic IV regimen in participants with AChR+ gMG is currently active (An open-label study to investigate the clinical efficacy of different dosing regimens of efgartigimod IV in patients with generalized myasthenia gravis [ADAPT NXT]; ClinicalTrials.gov identifier: NCT04980495).46 Moreover, another phase III, randomized, open-label study to assess the pharmacodynamic non-inferiority of subcutaneous versus IV administration of efgartigimod has just been completed (Evaluating the pharmacodynamic noninferiority of efgartigimod PH20 SC administered subcutaneously as compared to efgartigimod administered intravenously in patients with generalized myasthenia gravis [ADAPTsc]; ClinicalTrials.gov identifier: NCT04735432).47 Topline results show that the primary endpoint was met, with a comparable mean reduction in total IgG with the subcutaneous versus the IV formulation. Moreover, additional secondary endpoints were met, in particular 69.1% of patients were responders on the MG-ADL score and 65.5% of patients were responders on the QMG-score.47 From this, patients can enter an open-label study to receive efgartigimod 1,000 mg subcutaneously, to evaluate the long-term safety and tolerability of subcutaneous efgartigimod (Evaluating the long-term safety and tolerability of efgartigimod PH20 SC administered subcutaneously in patients with generalized myasthenia gravis [ADAPTSC+]; ClinicalTrials.gov identifier: NCT04818671).48 Finally, recruitment is currently underway for a phase II/III, uncontrolled, open-label trial investigating the safety, pharmacodynamics and pharmacokinetics of IV efgartigimod in children (2–18 years of age) with gMG (Evaluating the pharmacokinetics, pharmacodynamics, and safety of efgartigimod administered intravenously in children with generalized myasthenia gravis; ClinicalTrials.gov identifier: NCT04833894).49
A summary of all clinical trials of efgartigimod in gMG is provided in Table 1.26,43,44,46–50
Efgartigimod in other autoimmune diseases
Given its mechanism of action and pharmacodynamics, the potential benefits of efgartigimod have been explored in other autoimmune disorders. In patients with immune thrombocytopenia scarcely controlled by standard-of-care therapy, a phase II, randomized, double-blind, placebo-controlled study demonstrated that efgartigimod had a favourable safety profile and rapidly reduced total IgG levels, associated with clinically significant increases in platelet counts and, therefore, fewer patients with bleeding.38 These beneficial effects were maintained among patients who entered the OLE.38 A randomized, phase III, double-blind, placebo-controlled study assessing the efficacy and safety of IV efgartigimod in immune thrombocytopenia has just been completed, with positive results demonstrating that a higher proportion of patients treated with efgartigimod achieved a sustained platelet count response compared with placebo (A study to assess the efficacy and safety of efgartigimod in adult patients with primary immune thrombocytopenia [ITP] [ADVANCE]; ClinicalTrials.gov: NCT04188379).51 The OLE phase is currently ongoing (A long-term study to assess the safety and efficacy of efgartigimod in adult patients with primary immune thrombocytopenia [ITP] [ADVANCE+]; ClinicalTrials.gov identifier: NCT04225156).52 Also in immune thrombocytopenia, a phase III trial comparing the efficacy and safety of subcutaneous efgartigimod versus placebo is on-going (A study to evaluate the efficacy and safety of efgartigimod PH20 subcutaneous in adult patients with primary immune thrombocytopenia [ADVANCE SC]; ClinicalTrials.gov identifier: NCT04687072), while the OLE phase is currently recruiting (A phase 3 study to evaluate the safety and efficacy of efgartigimod PH20 subcutaneous in adult patients with primary immune thrombocytopenia [ADVANCE SC+]; ClinicalTrials.gov identifier: NCT04812925).53,54
In patients with mild-to-moderate pemphigus vulgaris or foliaceous, a phase II, open-label adaptive trial demonstrated an early effect of IV efgartigimod on disease activity and outcome parameters.39 Specifically, about 90% of patients achieved disease control (i.e. absence of new lesions and healing of previous established lesions) after a median of 17 days. Moreover, optimized prolonged treatment with efgartigimod combined with prednisone led to complete clinical remission in 64% of patients within 2–4 weeks.39 A phase III, randomized, double-blind trial evaluating the safety and efficacy of subcutaneous efgartigimod in pemphigus vulgaris or foliaceous is currently active (A study to assess the efficacy and safety of a subcutaneous formulation of efgartigimod PH20 SC in adults with pemphigus [vulgaris or foliaceus] [ADDRESS]; ClinicalTrials.gov identifier: NCT04598451), along with the OLE phase (A study to assess the long-term safety and efficacy of a subcutaneous formulation of efgartigimod PH20 SC in adults with pemphigus [vulgaris or foliaceus] [ADDRESS+]; ClinicalTrials.gov identifier: NCT04598477).55,56
The efficacy of subcutaneous efgartigimod versus placebo is currently being evaluated in a randomized, double-blind phase II trial in patients with chronic inflammatory demyelinating polyneuropathy (A study to assess the safety and efficacy of a subcutaneous formulation of efgartigimod in adults with chronic inflammatory demyelinating polyneuropathy [CIDP, an autoimmune disorder that affects the peripheral nerves] [ADHERE]; ClinicalTrials.gov identifier: NCT04281472), with the possibility for patients to enter the OLE assessing long-term efficacy and safety (A study to assess the safety and efficacy of a subcutaneous formulation of efgartigimod in adults with chronic inflammatory demyelinating polyneuropathy [CIDP, an autoimmune disorder that affects the peripheral nerves] [ADHERE+]; ClinicalTrials.gov identifier: NCT04280718).57,58 Finally, the BALLAD study, investigating the efficacy and safety of subcutaneous efgartigimod in bullous pemphigoid, is recruiting (A phase 2/3 study of efgartigimod PH20 SC in adult participants with bullous pemphigoid [BALLAD]; ClinicalTrials.gov identifier: NCT05267600),59 while the clinical study design to evaluate subcutaneous efgartigimod in myositis (ALKIVIA study) is being finalized.60
Final considerations
Given its innovative mechanism of action, efgartigimod lends itself to being used in many different autoimmune disorders. The safety and efficacy of efgartigimod in gMG have been confirmed in a randomized, double-blind phase III study,26 and on-going trials are evaluating its efficacy as different IV regimens and for subcutaneous administration.47,48 The positive results obtained so far further encourage a more targeted approach in MG, for which there is an urgent need. The algorithm for treating MG has remained the same for many years, relying mainly on non-selective immunosuppression, which comes with various burdensome side effects and greatly impacts patients’ quality of life. Moreover, up to 20% of patients with MG are refractory to conventional immunosuppressive therapy, meaning that they are often put on a high dosage of oral steroids and various NSISTs, without actually having a clinical benefit. In these patients, the burden is doubled, as patients are limited by both the disease and the side effects of chronic non-selective immunosuppression. Novel therapeutic approaches, including efgartigimod, may solve problems imposed by current therapy.
Real-world data collected after the FDA approval and from the early-access (or expanded-access in the US) programme will help to show how much efgartigimod will impact future MG treatment, in terms of reducing or even discontinuing steroids and NSISTs. Moreover, if the safety and efficacy of efgartigimod are confirmed, in future, its clinical benefit as a starting immunosuppressant for treatment-naïve patients can be explored, particularly in those patients who cannot start a steroid treatment for medical reasons, thereby continuously shifting the aim towards ‘precise’ medicine.
It is important, however, to underline that even if efgartigimod and other innovative therapeutic molecules hold great promise in replacing prednisone and NSISTs, their future prescription could be limited by their high costs, especially considering the need for life-long treatment.


