




Myasthenia gravis (MG) is an autoimmune disorder caused by autoantibodies against the myoneural junction, which lead to impaired neuromuscular transmission. These antibodies act at the post-synaptic membrane, commonly against the nicotinic acetylcholine receptor (AChR) but in some cases, antibodies to muscle-specific tyrosine kinase (MuSK) or lipoprotein receptor-related protein 4 (LRP4) can be seen. In an even smaller subset of cases, antibodies are not demonstrated on conventional antibody assays and these patients are referred to as ‘seronegative’.1
Patients with MG typically present with fluctuating muscle weakness usually associated with muscle fatigue (worsening weakness).2 Patients usually present with ocular features first, such as asymmetrical fatigable ptosis with or without double vision. The majority progress further and develop generalized muscle weakness involving the facial and pharyngeal muscles, the neck and axial muscles, and the limbs. Myasthenic crisis marks the severe end of the disease spectrum, which manifests with dysphagia that progressively worsens to complete loss of swallow function, often leading to respiratory muscle paralysis and type 2 respiratory failure.3 Myasthenic crisis is an emergency that requires prompt management in an intensive care unit.
Therapies include symptomatic treatment (acetylcholinesterase [AChE] inhibitors), immunomodulatory therapy (immunoglobulins and plasmapheresis), thymectomy and immunosuppressants, including corticosteroids and other steroid-sparing agents (Table 1).4 These traditional treatments can only bring about disease remission but are not curative. Moreover, 10–20% of patients with MG do not respond to current treatment.5 Patients who are MuSK antibody-positive respond less favourably to conventional treatment compared with patients who are AChR antibody-positive.6,7 Hence, there is a pressing need for newer and more effective immunotherapies for refractory MG.
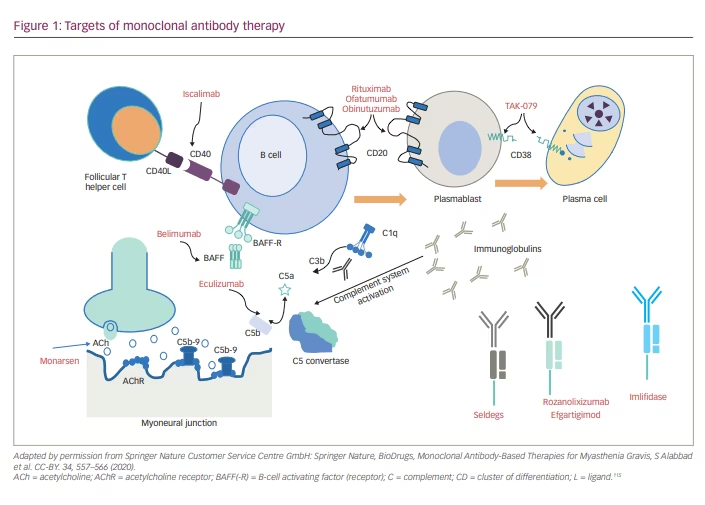
This review explains in detail the targeted therapies currently available and in development for MG. These therapies are classified as complement inhibitors, fragment crystallization (Fc) receptor antagonists, B-cell depletors, proteasome inhibitors, chimeric antigen receptor T-cell (CAR T) therapy, autologous stem cell transplantation and antisense oligonucleotide (Figure 1). These treatments promise rapid onset of action, fewer side effects, and more tolerability and sustained remission than conventional immunosuppressants. Thus, the newer drugs claim to improve the quality of lives of people with MG.
Corticosteroids: double-edged swords
Corticosteroid therapy was the first widely used immunosuppressive therapy introduced in MG. Steroids comprise the mainstay of immunosuppressive treatment in individuals with mild-to-moderate MG to induce remission.3 Around 80% patients on prednisolone achieve symptomatic remission.4 They are the most effective agents in treating MG. The onset of action is rapid with steroids, and they reduce the risk of progressing from ocular MG to generalized MG.8 However, their long-term administration is precluded by side effects, which include diabetes, hypertension, weight gain, peptic ulcers, accelerated bone loss, neuropsychiatric disturbances, cataracts, raised intraocular pressure and increased risk of infections.4
Animal models in myasthenia gravis
Animal models of experimental autoimmune myasthenia gravis (EAMG) can be induced by immunizing animals with Torpedo californica AChR in complete Freund’s adjuvant.9,10 The major histocompatibility complex class II genes influence the cellular and humoral immune response to AChR and lead to clinical EAMG developing in mice. Although EAMG can be induced in any animal species, most of the models are established in rats and mice because of the high incidence of clinical EAMG signs.11–13 A passive transfer of AChR antibodies can also induce EAMG.14 Despite the availability of EAMG models, except eculizumab, none of the currently used drugs has emerged from preclinical work in animal models.15,16
Strengths of animal models
The EAMG models have aided understanding of the role of autoantibodies and T helper (Th) cells. EAMG can be induced in mice and rats of susceptible strains that exhibit clinical features mimicking human MG. This helps in understanding both the muscle and the immune compartments to evaluate new treatments.17 EAMG models have AChR antibodies in serum, and deposits of complement and immunoglobulins at the neuromuscular junction, both of which mimic human MG pathology.11 Newer models using different pathophysiology, such as active MuSK EAMG, will help in simulating human MuSK MG.
Limitations of animal models
Disease in EAMG models does not occur spontaneously, and the induction procedure can significantly affect EAMG models by causing animal suffering and deaths, which also affects the statistical power of the study. Absence of thymic pathophysiology in animal models is a major limitation. Additionally, animal models are not able to simulate the role of environmental agents such as viral or bacterial organisms, which have an important role in the pathogenesis of autoimmunity.17
Treatment-refractory myasthenia gravis
Currently, there is no universally accepted definition of ‘treatment-refractory MG’. A few operational definitions are being employed for entry into clinical trials. Some of the characteristics of refractory MG are as follows.18–21
- Disease that does not respond to conventional immunosuppressive agents, used in adequate dose and duration, with persisting myasthenic symptoms or medication adverse effects that limit function.
- Need for on-going rescue treatment with intravenous immunoglobulin (IVIG) or plasmapheresis, while on immunosuppressive treatment.
- Intolerable side effects or comorbid diseases that preclude the use of conventional immunosuppressants.
- Frequent myasthenic crisis, even on immunosuppressive treatment.
There is no consensus on the number, dose and duration of immunosuppressants. Conventionally, if the patient does not respond to two or more immunosuppressants including steroids after ‘adequate’ dose and duration, they are considered refractory.
Pathophysiology of myasthenia gravis
MG is an autoimmune disorder driven by T lymphocytes, and it is modulated by a very sophisticated interaction between cluster of differentiation (CD)4+ T cells and B cells.22 The immune tolerance mechanism is disrupted in MG. The autoreactive T cells escape destruction in the thymus in the early part of life, leading to defects in central tolerance, and there is also a functional defect in regulatory T cells in peripheral blood, leading to reduced apoptosis of autoreactive T cells.23 The autoimmune mechanism begins when an infectious trigger disrupts the immune tolerance resulting from molecular mimicry between the infectious agent and the AChR.24 The antigen-presenting cells present the AChR to the CD4+ T cells leading to the release of multiple inflammatory molecules, such as interleukins (IL) and tumour necrosis factors.25 Regulatory T-cell (Treg) dysfunction leads to upregulation of Th1, Th2 and Th17 T cells that stimulate the production of antibody-synthesizing plasma cells and memory B cells.26–28 Plasma cells secrete antibodies of the immunoglobulin (Ig)G1 and IgG3 subclass.29,30 The antibody fragment binding site (Fab) binds to the AChR, and the Fc region attaches to the Fc receptor of the immune cells, bringing about the immune cascade.31
The integrity of the neuromuscular junction depends on effective AChR clustering and interaction of other proteins such as MuSK, LRP4, agrin and rapsyn.32 Antibodies against these proteins disrupt AChR clustering and bring about defective neuromuscular transmission, leading to muscle weakness.33
AChR antibodies and anti-LRP4 antibodies belong to the IgG1 subclass, whereas MuSK antibodies are IgG4. Short-lived plasma cells produce IgG4 (MuSK antibodies), whereas long-lived ones produce IgG1 and IgG3 (AChR antibodies and LRP4 antibodies). CD20 marker is predominantly present in short-lived plasma cells and not in long-lived plasma cells.
The AChR–antibody interaction activates the classical complement cascade and leads to the formation of the membrane attack complex (MAC).34 Calcium ions enter the MAC, resulting in membrane damage and release of AChR-containing membrane debris into the synaptic space.33 Thus, the damaged membrane leads to reduced response to acetylcholine and, therefore, impaired myoneural transmission.
The pathogenesis is different in MuSK antibody-mediated disease because the MuSK antibody belongs to the IgG4 subclass and, therefore, does not stimulate the complement cascade.35 Antibodies against MuSK reduce interaction between MuSK and LRP4, leading to defective AChR clustering and impaired neuromuscular transmission. Anti-LRP4 antibodies are of IgG1 subclass and, hence, activate the complement system.36
The advances in knowledge about the various immune processes in MG have led to the development of drugs that can selectively antagonize the immune cascade involved in MG.
Novel molecular therapies in myasthenia gravis
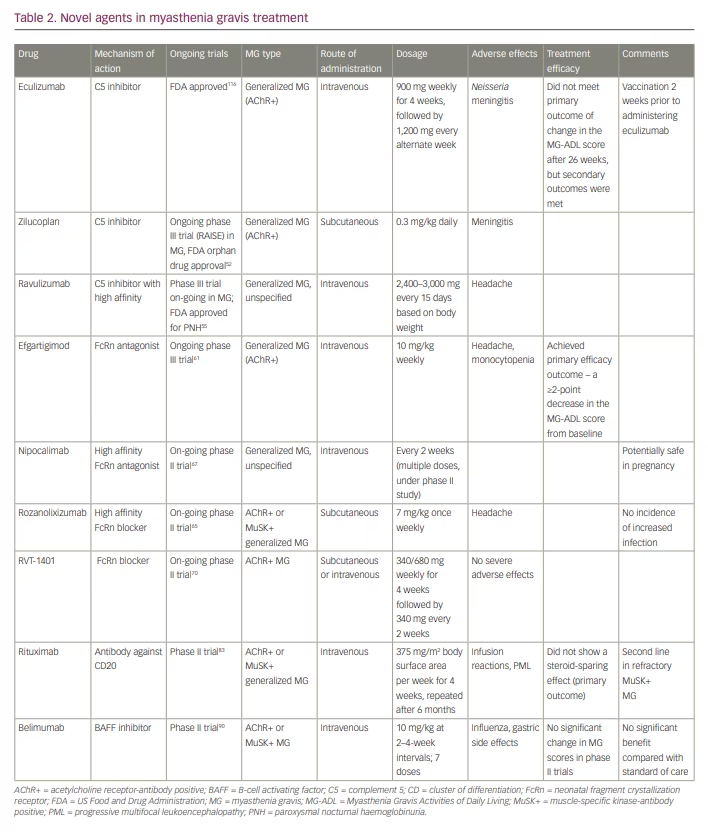
These agents are broadly classified as complement inhibitors, Fc receptor antagonists, B-cell depleting agents (anti-CD19 and CD20, and B-cell activating factor [BAFF] inhibitors), proteasome inhibitors, T cells and cytokine-based therapies (CAR T-cell therapy), autologous stem cell transplantation and antisense oligonucleotide. Most of these treatments differ from the conventional immunosuppressants because they have a faster onset of action, favourable side-effect profile, and can potentially maintain long-term remission.22 Table 2 summarises the various novel immunotherapeutics in MG.
Complement inhibitors
Once the IgG antibody binds to the AChR receptor, it stimulates the classical and common complement pathways. First, complement 5 (C5) convertase is formed, which splits into C5a and C5b, and C5b then combines with C6–C9 complement proteins to form the MAC. The MAC formed then incorporates into the plasma membrane and leads to cell lysis.37 Thus, the post-synaptic membrane shows the presence of complement deposits and IgG.
Therefore, the complement inhibitors increase the AChR content in the post-synaptic membrane by preventing the MAC being formed. Many animal models have shown reduced MAC deposition at the post-synaptic membrane in spite of increased IgG levels and a concomitant improvement in muscle power.38,39
Eculizumab
Eculizumab is the first available drug that blocks the C5 protein of the complement cascade.40 Eculizumab is a recombinant monoclonal antibody that binds to C5 and blocks its conversion into C5a and C5b, thereby preventing the formation of MAC.41 The drug is approved by the US Food and Drug Administration (FDA) for the treatment of paroxysmal nocturnal haemoglobinuria (PNH), generalized MG, atypical haemolytic uraemic syndrome and neuromyelitis optica.42,43
The major benefits of this drug have been demonstrated in a large phase III trial (REGAIN).44 In REGAIN, 126 patients with refractory generalized MG were randomized to either eculizumab or placebo as an add-on treatment to on-going immunosuppressive treatment for a period of 26 weeks. The induction dosage was 900 mg intravenously on days 0, 7, 14 and 21 (1,200 mg on day 28); and the maintenance dosage was 1,200 mg every second week for 26 weeks. The primary efficacy endpoint was a change in the Myasthenia Gravis Activities of Daily Living (MG-ADL) score.
The primary outcome was not attained, but secondary outcomes, such as change in MG-ADL, Quantitative Myasthenia Gravis (QMG) score and Myasthenia Gravis Quality of Life 15-item Scale (MG-QoL15) scores showed significant improvement in the eculizumab group.44 Eculizumab does not have any role in reducing antibody production and acts only on the complement cascade.45
The risk of meningococcaemia is another potential problem with eculizumab, and patients receiving this drug should be vaccinated against Neisseria meningitidis 2 weeks prior to starting eculizumab.46 The beneficial effects of eculizumab compared with placebo led to its approval by the FDA for its use in refractory, AChR antibody-positive MG.47 An open-label extension of the REGAIN trial also demonstrated a significant reduction in rates of exacerbation of MG, rates of MG-related hospitalization and increase in global clinical improvement scores.48 More than 50% of patients achieved minimal manifestation (MM) status or pharmacological remission in the open-label extension study.
Though modest, the success of eculizumab has paved the path for developing other complement-inhibiting molecules. However, a major concern is that complement inhibitors do not alter the primary immunopathology and the need for concomitant immunosuppression is still inconclusive.36
Zilucoplan
Zilucoplan is a synthetic macrolide that has the same mechanism of action as eculizumab.22 Zilucoplan prevents the downstream MAC formation by attaching to C5 and blocking conversion to C5a and C5b.49 The binding site of zilucoplan is different from that of eculizumab, and zilucoplan can be employed for patients unresponsive to eculizumab.50
This has been tested in a phase II randomized controlled trial in which two doses of subcutaneous zilucoplan were compared in patients with AChR antibody-positive, moderate-to-severe, generalized MG with a QMG score ≥12.51 Patients were randomized 1:1:1 to subcutaneous doses of 0.1 mg/kg, 0.3 mg/kg and placebo, for 3 months. The 0.3 mg/kg group had more statistically significant and clinically meaningful effect compared with the placebo, favouring zilucoplan. The response noticed with the 0.1 mg/kg group was also statistically significant; however, the improvement was noticed 1 week after treatment onset. No serious adverse effects were noted. A phase III randomized controlled trial (RAISE) is on-going and includes patients with non-refractory MG
as well.52
Ravulizumab
Ravulizumab is a humanized monoclonal antibody that has the advantage of a longer half-life compared with eculizumab because of a different amino acid substitution at the Fc region, thereby providing a sustained reduction in C5.53 Ravulizumab can be administered once every 2 months, with a single loading dose administered on day 1 and maintenance dose on day 15. This drug is FDA approved for treating PNH.54 A phase III randomized controlled trial that began in 2019 is on-going in patients with generalized MG.55 The safety profile matches that of eculizumab.
Neonatal Fc receptor blockers
Another way to target the immune pathway in MG is to reduce the autoantibody circulation. The neonatal Fc receptors (FcRn) help protect IgG from lysosomal degradation and thus prolong the half-life of immunoglobulins.56 Blocking the FcRn will reduce IgG recycling.57 The rapid fall in IgG levels following administration of FcRn antagonists is not accompanied by a parallel increase in IgG synthesis.58 Therefore, FcRn antagonists are an attractive treatment target in MG. On-going trials are using different molecules, described in detail below. These agents have not been tried in seronegative patients with MG, and longer treatment durations are necessary to confirm the efficacy and safety of these agents.
Efgartigimod
Efgartigimod is a humanized IgG1 Fc portion of antibody with great affinity for FcRn.59 A double-blind, placebo-controlled phase II trial over a 3-week period performed in 24 patients with AChR antibody-positive MG revealed a 50% reduction in IgG concentration at the third week, and levels returned to normal after the last dose.60 This trial did not include patients with Myasthenia Gravis Foundation of America (MGFA) class I (restricted ocular MG), class IVB (bulbar involvement) and class V (myasthenic crisis) patients. The most common side effects noted were headache and reduced monocyte counts, and a single case of shingles was also reported. There were improvements in all the MG scales used in the study, and these changes paralleled the drop in IgG levels, but persisted even after the IgG levels returned to baseline.
The phase III ADAPT trial aimed to assess the safety and efficacy of efgartigimod in patients with generalized MG.61 Adults with MGFA class II–IV and MG-ADL score of at least 5, currently treated with a stable dose of at least one MG drug, were studied. Overall, 167 patients entered the trial, 84 received efgartigimod and 83 received placebo. The primary efficacy outcome was achieved; 44 (68%) of 65 patients with AChR antibody-positive MG in the efgartigimod group gained a clinically meaningful improvement, at the end of 10 weeks. A clinically meaningful improvement was considered as ≥2-point decrease in total MG-ADL score from baseline, compared with 19 (30%) of 64 patients in the placebo group (odds ratio 4.95, 95% confidence interval [CI] 2.21–11.53, p<0.0001) at the end of cycle 1 . The most common adverse events were headache and nasopharyngitis.61
Rozanolixizumab
Rozanolixizumab is a humanized anti-FcRn monoclonal IgG4 antibody.62 A phase I, placebo-controlled, randomized safety trial evaluated healthy participants, who were randomized to receive a single infusion of 1 mg/kg, 4 mg/kg or 7 mg/kg intravenous or subcutaneous rozanolixizumab, or placebo.63 The reduction in IgG concentration following rozanolixizumab administration peaked at 7–10 days and IgG returned to baseline levels by day 57 across the three arms. Side effects, such as headache, vomiting, nausea and fever, were encountered more with the intravenous route than the subcutaneous route.63 A subsequent phase IIa trial enrolled 43 patients with generalized MG who were randomized to receive three once-weekly subcutaneous infusions of placebo or rozanolixizumab, and were re-randomized 4 weeks later to three once-weekly doses of either 4 mg/kg or 7 mg/kg rozanolixizumab.64 Benefits were noted in all the MG scores employed in the treatment arm. However, headache was more frequent in the rozanolixizumab arm, and 16% patients withdrew from the study. Phase III trial is currently underway.65
Nipocalimab
Nipocalimab is a completely humanized, IgG1, anti-FcRn antibody with a high specificity for FcRn at both endosomal and extracellular pH.59 A randomized, double-blind, placebo-controlled first-in-human study was conducted in 50 healthy adults.66 Multiple weekly doses of 15 or 30 mg/kg of nipocalimab led to a mean reduction in the levels of IgG of about 85% from baseline, which was maintained at 75% from baseline for up to 24 days. It was well tolerated. A phase II trial is on-going.67 Nipocalimab is not expected to cross the placenta68 and its safety in pregnant women needs to be further studied.
RVT-1401
RVT-1401 is a humanized monoclonal antibody that can be administered intravenously or subcutaneously. In a phase I trial, healthy individuals were randomized into two cohorts− single-dose cohort and multiple ascending doses cohort.69 The initial group was given 0.1 mg/kg intravenously (n=4) or 0.5 mg/kg subcutaneously (n=3) doses. The subsequent cohorts were randomized in such a way so as to receive single subcutaneous or intravenous doses of RVT-1401 or placebo (6:2), the dose ranging from 100 to 765 mg. Individuals in the multiple ascending dose group had received four weekly subcutaneous doses of RVT-1401 (340 mg or 680 mg) or placebo (8:2). RVT-1401 was well tolerated and its administration reduced the plasma IgG concentrations following both intravenous and subcutaneous routes. The drug is being tested in a phase II trial in patients with AChR antibody-positive MG.70
Limitations of complement inhibitors and neonatal Fc receptor antagonists
Complement blockers and FcRn antagonists are novel agents that promise to increase the efficacy and safety of AChR antibody-positive MG treatment in the near future; however, there are a number of shortcomings with these agents, including the following.
- The studies have not included seronegative patients with MG.
- The long-term efficacy of these agents and their adverse effects are not well characterized; hence, studies with longer treatment durations are necessary.
- The use of these novel agents in myasthenic crisis is not studied.
- The place of these agents in the MG treatment algorithm is also uncertain.
- Inhibiting FcRn can alter serum levels of therapeutic monoclonal antibodies, and the pharmacokinetic interactions among these agents need to be studied in detail.59
- The need for concomitant immunosuppression with complement inhibitors is unclear, especially in patients where primary pathogenesis is not addressed.
B-cell inhibitors
B cells have a key role in the pathophysiology of MG.71 The pathogenic autoantibodies are produced by plasma cells and plasmablasts are formed from B cells. Various molecular and cellular factors are involved in stimulating the proliferation of autoreactive B cells, among which BAFF is the most important.72 In addition to producing antibodies, B cells also present antigens to T cells, and activate and upregulate a pro-inflammatory state. Thus, B-cell depletors (biologic agents against CD19- and CD20-postitive B cells) have a major role in MG treatment.
Rituximab
Rituximab has gained wide popularity in the management of MG, and was initially employed for treating cancer and connective tissue diseases.73–76 Rituximab is a murine−human chimeric monoclonal antibody against CD20 (a transmembrane protein produced by B cells), and acts by multiple mechanisms.77 It directly produces complement-mediated cytotoxicity and antibody-dependent, cell-mediated cytotoxicity. Rituximab also causes structural changes in the B cells, and promotes apoptosis. Moreover, rituximab also increases the Treg cells, which helps in slowing down the progression of disease symptoms, reducing the need for other drugs.
In a systematic review, 99 patients with AChR antibody-positive MG and 57 patients with MuSK antibody-positive MG treated with rituximab were evaluated for safety and efficacy of rituximab. It revealed that 44% of patients attained MGFA MM status or better. Complete pharmacological and medical remission was attained in 27% of patients. Patients who were MuSK-antibody positive had better responses and fewer relapses compared with patients who were AChR-antibody positive. In this study, 72% of MuSK antibody-positive patients attained MM status or remission, compared with 30% of AChR antibody-positive patients. Younger age of disease onset and milder disease also predicted a favourable response. The study did not show any correlation between the reduction in antibody levels and clinical response to rituximab.78
A multicentre study conducted in MuSK antibody-positive patients with MG also showed better response to rituximab compared with standard immunosuppressants.79 An Austrian retrospective study also revealed, at a median follow-up of 20 months, patients with MG (70% AChR-antibody positive, 25% MuSK-antibody positive) on rituximab achieved remission in about 43% of cases and MM status in 25% of cases.80 MuSK antibody-positive patients attained remission more frequently than AChR antibody-positive patients (71% versus 36%, respectively). A retrospective review from Stockholm showed the superiority of rituximab in shortening the time to disease remission and reducing the need for additional immunosuppressive treatment in patients with new-onset MG. The time to remission was shorter with rituximab compared with those who received other immunosuppressive therapy.81
A randomized controlled trial compared rituximab with placebo as add-on treatment in patients with AChR antibody-positive generalized MG (the BEAT-MG study).82 The study included 52 patients on a stable prednisone regimen for 4 weeks or prednisone plus another immunosuppressive treatment for 6 months. Rituximab or placebo was administered in two cycles, 6 months apart. The primary outcome was a steroid-sparing effect (≥75% reduction in mean daily prednisone requirements in the 4-week period before week 52 compared with the 4-week period before randomization). Rituximab did not have a steroid-sparing effect compared with placebo (60% on rituximab versus 56% on placebo).83 Also, no significant differences in disease-severity outcomes were noted. The trial result shows that in mild-to-moderate AChR antibody-positive MG, rituximab is unlikely to produce a clinically meaningful steroid-sparing effect over 12 months. However, there are data from large observational prospective studies that support use of rituximab.18,84 Hence, there is an urgent need for an adequately powered phase III randomized controlled trial with rituximab, which includes both AChR and MuSK antibody-positive patients. There are few cases of progressive multifocal leukoencephalopathy reported with rituximab infusion.85
Other B-cell inhibitors include the following.
- Ofatumumab: humanized anti-CD20 antibody, commonly used in refractory rheumatoid arthritis.86 This drug was used in a single patient with MG who did not respond to rituximab.87
- Iscalimab: anti-CD40 monoclonal antibody, used as an add-on treatment in moderate-to-severe AChR antibody-positive MG.88
- Belimumab: an IgG1l(lambda) monoclonal antibody against B-lymphocyte stimulator or BAFF, approved for use in systemic lupus erythematosus as a BAFF inhibitor.89 It was tested in a phase II trial in patients with AChR antibody-positive generalized MG, but it did not show a meaningful change in MG scores.90
- Tocilizumab: humanized monoclonal antibody against the IL-6 receptor.91 It induced improvement in two patients with refractory MG with rituximab failure.92
- Obinutuzumab: humanized anti-CD20 monoclonal antibody. It was administered in a patient with MG and chronic lymphocytic leukaemia (CLL); there was complete disease remission of CLL, and MG was undetectable after treatment.93
Proteasome inhibitors
The plasma cells are the differentiated B cells, and they have lost cell-membrane markers and are therefore resistant to most immunosuppressive treatments in healthy adults.94 The plasma cells form sentinels of adaptive immunity, and thus contribute to treatment resistance in MG. Plasma cells continuously produce antibodies and are sites of increased protein turnover. For effective cellular homeostasis, these proteins need to be destroyed and removed, which is carried out in proteasomes. Inhibiting proteasomes leads to misfolded protein accumulation and plasma-cell apoptosis, thus reducing the antibody burden.95
Bortezomib, a proteasome inhibitor, was employed in a patient with treatment-refractory MuSK antibody-positive MG. There was moderate improvement in myasthenic symptoms, but the patient had already been treated with rituximab 19 days before receiving bortezomib, which is a major confounding factor.96 More trials are needed to assess the efficacy of bortezomib in MG.
Anti-T-cell and cytokine-based therapies
As Treg cell dysfunction and Th1 and Th2 overactivity are encountered in MG,97 agents that modulate T cells are promising in MG treatment.39 Numerous monoclonal antibodies have been manufactured against T cells and cytokine pathways, including secukinumab (an IL-17A inhibitor), rontalizumab (an INFα inhibitor) and tocilizumab (an IL-6 inhibitor).
Chimeric antigen receptor T-cell therapy
The CAR T cells are genetically engineered autologous T lymphocytes that, when infused into the patient, recognize tumour antigens and induce tumour cell death.98 This concept is being employed in cancer therapy; mainly in refractory B-cell acute lymphoblastic leukaemia (ALL), non-Hodgkin lymphoma and B-cell lymphoma.99,100 However, major adverse effects of CAR T-cell therapy is cytokine release syndrome.101 Studies have shown effectiveness in certain autoimmune conditions such as lupus and pemphigus, and thus, CAR T-cell therapy is a potentially attractive option in MG.102,103 The currently on-going, phase I/II, Descartes-08 trial is using CD8-positive CAR T-cells directly against plasma cells expressing B-cell maturation antigen in patients with MG.104,105
Autologous haematopoietic stem-cell transplantation
Autologous haematopoietic stem-cell transplantation (HSCT) has been tested in a series of seven patients with severe MG (MGFA class III–IV). In this Canadian retrospective series, all patients were in stable remission after a median follow-up of 40 months, and the authors suggested that HSCT might result in remission of MG for a long duration.106 HSCT is thought to reset the immune system, and it also leads to avoidance of maintenance immunosuppressive treatment.106 With safer induction regimens, HSCT can be a safe and attractive treatment for severe MG in future.
Antisense oligonucleotide (monarsen)
The use of antisense oligonucleotides in treating MG is mainly based on the research conducted by Soreq et al. at the Hebrew University in Israel.107 These agents act by selectively hybridizing with AChE messenger RNA (mRNA); this activates ribonucleases that destroy the mRNA–antisense complex and stop the protein synthesis.108 The antisense oligonucleotide monarsen (EN101) acts on exon 2 of AChE mRNA and makes it more susceptible to destruction, thereby maintaining the acetylcholine levels in the synaptic cleft.108 Phase IIa studies showed some improvement in QMG scores, and good drug tolerability,109,88 but currently, no phase III trials are testing the efficacy of monarsen.
Future potential therapies
Seldegs
These are a novel class of agents that have considerable potential in antibody-mediated autoimmune diseases. The knowledge that the FcRn maintains IgG levels and transport in the body has fuelled the development of antibody- or peptide-based inhibitors to reduce IgG levels. FcRn blockers inhibit the interaction of the Fc region of IgG with FcRn, and decrease levels of IgGs of all specificities, even protective antibodies.110 To overcome these off-target effects, the ‘seldegs’ (selective degradation) were developed. These engineered antibodies selectively clear antigen-specific antibodies without modulating the levels of antibodies of other specificities.110 These agents have been tried in myelin oligodendrocyte glycoprotein antibody-related demyelination in experimental animals.111 Thus, these are promising future therapeutic agents in treating MG.
Imlifidase
Imlifidase, derived from Streptococcus pyogenes, is an endopeptidase enzyme that specifically cleaves IgG and has been used in patients before renal transplantation.112 This drug may also be suitable for the treatment of MG, although its immunogenicity is likely to limit its repeated use.113
Preclinical studies of newer drugs in myasthenia gravis
Other than eculizumab, most of the currently used drugs have not been studied in preclinical studies, even though the pathophysiology of MG is well established and there is availability of reasonably good animal models.17 With the surge in monoclonal antibodies in the autoimmune field, the importance of well-conducted preclinical studies has increased to avoid failure of large clinical trials. As in other fields, such as stroke neuroprotection, it is recommended that the quality of preclinical trials improves to that of human clinical trials.114
A summary of various improvement measures is given below.115
- Ensure animal models include both sexes.
- Include animals with minimum degree of weakness (e.g. Grade II power according to the MRC grading).
- Randomize treatment allocation in animals to control for known and unknown confounding factors.
- Always use blinded outcome assessments in any subjective endpoint in animal studies.
- Adequately power preclinical studies, and calculate sample size.
- Pre-register preclinical study protocols to help avoid publication bias.
- Ensure preclinical studies are replicable by independent researchers.
- Test potential drugs in animal models after the animals manifest the disease.
- Use and develop better markers for disease activity and progression, as outcome parameters in preclinical studies do not match those used in clinical trials.
- Use different preclinical models representing different clinical and pathological subtypes (e.g. ocular versus generalized MG; AChR- versus MuSK-antibody positive).
- Use globally acceptable reporting guidelines similar to CONSORT guidelines for clinical trials.
Conclusion
More focused immunotherapies offer better treatment options for patients with MG. Rapid onset of action, sustained disease remission and favourable side-effect profiles of the novel treatments are attractive. However, many drugs are not meeting their primary endpoints, and their long-term safety is unknown. Better preclinical studies are required to select drugs to be tested in humans. More randomized controlled trials are needed, as well as head-to-head trials among the newer immunotherapies, trials investigating different dosing regimens (early versus late), and studies into seronegative MG and the role of concomitant immunosuppressants.


