




POEMS syndrome is a rare paraneoplastic syndrome characterized by a complex constellation of systemic findings in the setting of a proliferative cell disorder. Named for peripheral neuropathy, organomegaly, endocrinopathy, M-proteins, and skin changes, these findings are predominant among several other associated features of the syndrome.
We describe a case in which the diagnosis of POEMS was suspected in the setting of neuropathy with skin changes, and was confirmed by subsequent hematological evaluation. Special emphasis in this case report is placed on the neurological clinical suspicion and workup in identifying a life-threatening condition, particularly the clinical and diagnostic challenge of distinguishing POEMS from chronic inflammatory demyelinating polyneuropathy (CIDP). Our experience with this patient reinforces the importance of early recognition and diagnosis of POEMS.
Case report
A 44-year-old previously healthy male presented to his primary care provider with 2 weeks of dull bilateral foot pain localized at the great toes and aggravated by movement. He denied any injury to the toe or erythema of the joint. Examination at that time was notable for absent sensation to light touch and monofilament over the distal parts of the feet. Radiographic imaging demonstrated mild bilateral hallux valgus deformities with small bunions, without any fracture, dislocation, or suspicious osseous lesion. At a subsequent visit, 3 months after the onset of symptoms, an electrodiagnostic study was requested. It revealed a sensory-motor neuropathy with length-dependent characteristics (Table 1, Table 2). Pathophysiology was consistent with a demyelinating process with secondary axonal loss. Diagnosis of CIDP was considered by the electromyographer.
The patient was subsequently lost to follow-up and presented 8 months later at the Neurology Clinic with progressive weakness and worsening distal sensory dysesthesias. Basic neuropathy workup for treatable causes was unremarkable. Cerebrospinal fluid (CSF) showed albuminocytologic dissociation with a protein of 98 mg/dL.
The patient was then referred to our Neuromuscular Clinic with a clinical diagnosis of CIDP and recommendation for intravenous immunoglobulin therapy (IVIG). Additional history obtained indicated an unintentional weight loss of 50 pounds over 8 months and intermittent headache with blurry vision over 3 months. Motor examination (based on the Medical Research Council Motor Examination scale of 0–5) revealed upper extremity strength 5; lower extremity: hip flexion 4, hip extension 5, knee flexion 5, knee extension 5, ankle dorsiflexion 2, and ankle plantarflexion 3. Sensation was decreased in a symmetric stocking-glove distribution to temperature, pain, and light touch to the lower third of the calves, and decreased vibration to the level of the knees. Reflexes were absent. Additional examination revealed bilateral disc edema and hyperpigmentation of the skin.
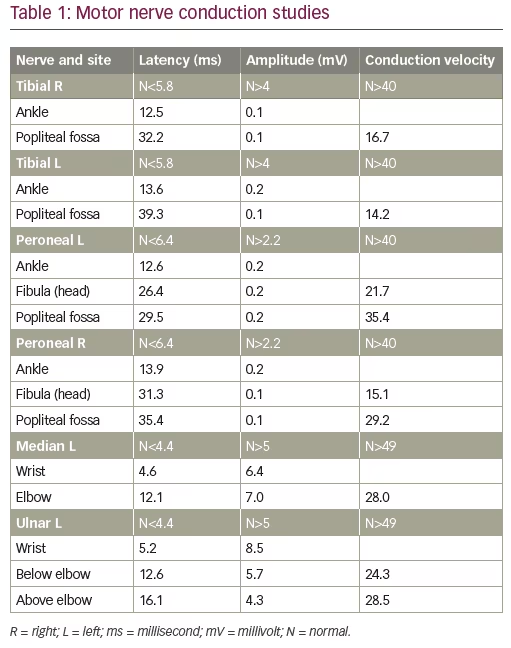
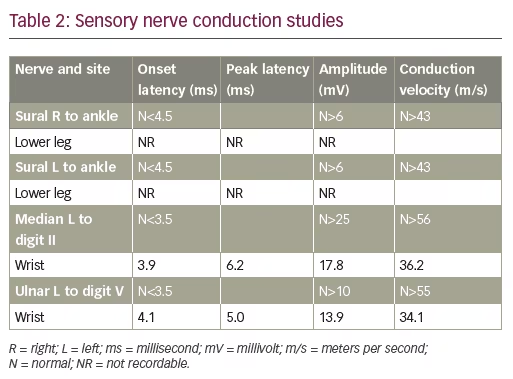
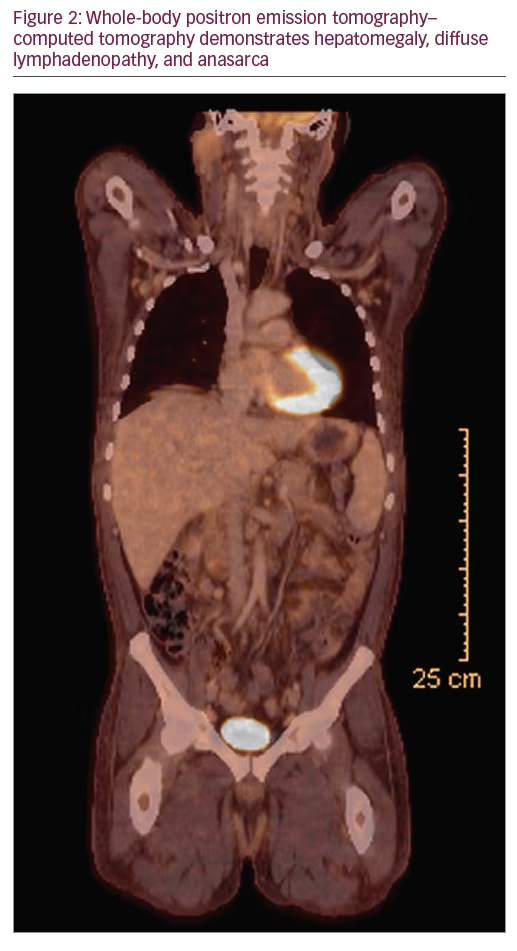
On that encounter, suspicion rose for an underlying process associated with malignancy, and a clinical diagnosis of POEMS was suspected. Further laboratory evaluation was notable for thrombocytosis. Immunofixation identified an IgA lambda monoclonal protein. Serum-free kappa/lambda light chain ratio was normal. Quantitative immunoglobulin levels revealed an elevated IgA 563 mg/dL (normal 79–455 mg/dL), with lambda light chain 115.3 mg/L (normal 5.7–26.3 mg/L) and kappa light chain 73.5 mg/L (normal 3.3–19.4 mg/L). Serum vascular endothelial growth factor (VEGF) was elevated to 332 pg/mL (normal <86 pg/mL). Nuclear medicine (NM) bone imaging with technetium 99m-methyl diphosphonate was performed and showed focal increased radiotracer uptake in the left T12 vertebral body (Figure 1), which corresponded to a sclerotic lesion on follow-up positron emission tomography–computed tomography (PET/CT). Multiple additional sclerotic lesions were identified in the manubrium, bilateral ribs, cervical, thoracic, and lumbar spine, posterior right acetabulum, and iliac bones bilaterally. PET/CT was also notable for hepatomegaly, as well as multiple prominent cervical, abdominal, pelvic, and inguinal lymph nodes measuring up to 1 cm. This was accompanied by anasarca, including diffuse mesenteric and perinephric edema (Figure 2).
![]()
The patient underwent core biopsies of the right acetabulum sclerotic bone lesion which showed a mild increase in CD138+ plasma cells comprising 5% of cellularity, however without definitive light chain restriction. Bone marrow biopsy showed normocellular bone marrow with moderate erythroid hyperplasia, and 9% plasma cells with lambda light chain restriction.
The patient was initiated on lenalidomide-dexamethasone therapy for POEMS. Two weeks after receiving his first dose, the treatment course was complicated by non-ST-elevation myocardial infarction. Lenalidomide was stopped at this point, and the patient was transitioned to CyBorD chemotherapy (cyclophosphamide, bortezomib, dexamethasone) due to concern for the progression of neuropathy.
Discussion
The constellation of peripheral neuropathy, organomegaly, endocrinopathy, M-proteins, and skin changes was first described as POEMS in 1980 by Bardwick et al in a collection of case studies.1 The findings were also separately described in case reports by Crow in 1956,2 Fukase in 1968,3 and Takatsuki and Sanada in 19834 as peripheral neuritis accompanied by diffuse pigmentation of the skin in the setting of myelomatosis and plasma cell dyscrasia with polyneuropathy and endocrinopathy, lending to its alternate names of Crow–Fukase and Takatsuki disease. Despite these case reports and multiple subsequent studies, the pathogenesis of POEMS syndrome is not well understood.5
Clinical symptoms
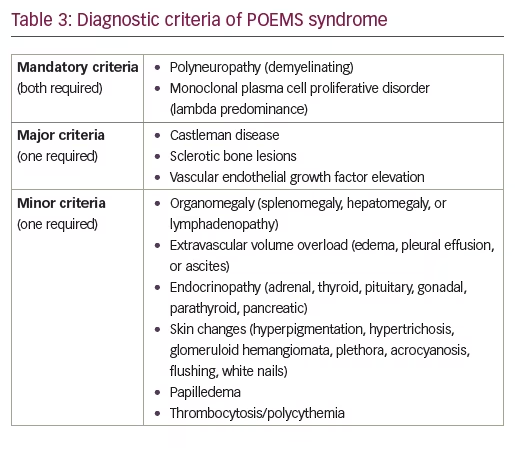
The diagnosis of POEMS requires a combination of clinical and/or laboratory findings, including two mandatory criteria plus one major and one minor criterion (Table 3): of note, all features of the acronym need not be present. Necessary for the diagnosis are polyneuropathy and monoclonal plasma cell proliferative disorder. Major criteria include the presence of Castleman disease, sclerotic bone lesions, or vascular endothelial growth factor elevation. Minor criteria include organomegaly, extravascular volume overload, endocrinopathy, skin changes (such as hyperpigmentation, hypertrichosis, glomeruloid hemangiomata, plethora, acrocyanosis, flushing, white nails), papilledema, thrombocytosis, and polycythemia.6 Nearly all of these features and laboratory findings were observed in the later stages of our patient’s disease.
Castleman disease is a rare lymphoproliferative disorder characterized by unicentric or multicentric7 angiofollicular lymph node hyperplasia, and is associated with the development of other malignancies8 as well as neuropathy. The Castleman disease variant of POEMS has greater severity of demyelinating features and degree of axonal loss when compared with Castleman disease-associated neuropathy that does not fit additional criteria for POEMS.9
Worse prognosis is portended by extravascular volume overload, pulmonary hypertension, respiratory symptoms, impaired diffusion capacity of the lungs for carbon dioxide (DLCO), fingernail clubbing, and papilledema.5 Papilledema is an early sign and is an independent adverse prognostic factor. The pathophysiology of papilledema in POEMS syndrome is contested,10 but it is associated with raised intracranial pressure, elevated CSF protein levels,10 and increased vascular permeability secondary to elevated serum VEGF.11
While the constellation of symptoms is more easily recognizable in advanced disease, it is important to have high clinical suspicion for POEMS when patients first present due to concern for underlying malignancy. Over 50% of patients with POEMS syndrome initially complain of neuropathy. In the setting of bilateral sensory and motor involvement, symptoms are often mistaken for CIDP and are only reconsidered when symptoms are refractory to IVIG therapy. In fact, over 60% of cases are initially misdiagnosed as CIDP12 and the median time to diagnosis is 13–18 months after onset of initial symptoms.13,14 Due to this delay in correct diagnosis, appropriate management with radiation, chemotherapy, or bone marrow transplant is often postponed with potential worsening of symptoms and progression of underlying systemic disease.
Diagnostic testing
Due to the similarities in clinical history and results of diagnostic testing in POEMS syndrome and CIDP, it is often challenging to make the correct diagnosis. Both CIDP and POEMS are chronic progressive neuropathies with motor and sensory components. Electrodiagnostic testing in both entities captures demyelinating polyradiculoneuropathy with axonal features. Although both POEMS and CIDP have significant slowing of conduction velocity, electrophysiological findings in POEMS syndrome show rare conduction block, suggesting that POEMS does not share the same immune-mediated focal demyelination as CIDP.15 Demyelination in POEMS predominantly involves the nerve trunk, where the nerve-blood barrier is vulnerable to breakdown in the setting of VEGF.12 In POEMS syndrome, there is more axonal loss than in CIDP, as evidenced by reduced amplitudes of distal motor and sensory potentials. Additionally, greater attenuation of the compound muscle action potentials affects the lower limbs more so than upper limbs in a length-dependent pattern.12,16 Wallerian degeneration in axonal polyneuropathy may result in terminally accentuated conduction slowing.15 CSF protein level does not differentiate between either condition. A prior study comparing sural nerve biopsies in POEMS and CIDP described that POEMS has higher rates of axonal degeneration, diffuse myelinated nerve fiber loss, uncompacted myelin lamellae, opening of tight junctions between endothelial cells, pinocytic vesicles adjacent to cell membranes, and increased number of small epineurial blood vessels (>120).17,18 In CIDP, biopsies reveal evidence of endoneurial inflammation, macrophage-associated demyelination, multifocal myelinated fiber loss, and onion bulb formation.12,16,17 There is, however, no need, except in rare situations, to perform a nerve biopsy for diagnostic purposes.
A retrospective review comparing POEMS and CIDP patients showed a statistically significant elevation in platelet count with POEMS, suggesting that it is an easy marker when considering the diagnosis of POEMS.19 Thrombocytosis has since been added to the list of diagnostic minor criteria, and as a readily available laboratory examination may prove to be a useful screening tool.
Treatment
The degree of recovery from POEMS neuropathy relies on prompt treatment.20 While IVIG and plasmapheresis are proven therapies for CIDP, they are ineffective in POEMS and may delay appropriate management. Treatment for POEMS targets the plasma cell clone and is dependent on the degree of plasma cell infiltration into bone marrow.6 Therapies are based on expert recommendation and case series, as there are no existing randomized clinical trials involving POEMS syndrome.
Isolated bony lesions (up to three) without diffuse bone marrow involvement are treated with a standard 40 Gy of radiation. Symptoms improve over 3–36 months, and treatment with radiation is potentially curative.21 At times, radiation is accompanied by adjuvant corticosteroid therapy for acute symptomatic management.20
If bone marrow is involved, systemic chemotherapy is required. Therapy mirrors that of multiple myeloma and light chain amyloidosis, typically involving high-dose alkylator-based chemotherapy with autologous peripheral blood stem cell transplantation (ASCT), or low-dose chemotherapy with corticosteroids. ASCT has demonstrated a response rate of 100% in case studies, with recommendations for ASCT with high-dose chemotherapy lenalidomide (15–25 mg orally days 1–21, every 28 days) and weekly dexamethasone. An alternate regimen is cyclophosphamide (750 mg/m2 intravenously every 3 weeks) and 4–5 days of corticosteroid therapy.20 Additional treatments include thalidomide or bortezomib, such as in our patient. Expected neurological response to systemic chemotherapy and ASCT is 6–36 months after completion of therapy.6 Of note, lenalidomide-dexamethasone combination carries the risk of thrombosis, while thalidomide and bortezomib are associated with peripheral neuropathy. VEGF levels are monitored as a surrogate of therapeutic response. However, anti-VEGF therapies such as bevacizumab that lead to undetectable VEGF levels disappointingly have not translated to consistently demonstrable clinical benefit.20
Conclusion
As patients with POEMS syndrome have an increased risk of life-threatening thrombogenic events including myocardial infarction, ischemic stroke, splenic infarction, and venous thrombosis, any mis-diagnosis as CIDP or delay in appropriate diagnosis may result in disease progression and additional complications. High clinical suspicion and recognition of the electrodiagnostic findings early in the disease course when a patient presents only with neuropathy are key in the proper diagnosis and management of POEMS. Cases of ‘CIDP’ or monoclonal gammopathy should be studied for the possibility of this complex syndrome.


